
Optetrak Patella Implant Device Exactech Recall
A letter was recently sent to surgeons, hospitals, and healthcare professionals announcing the voluntary recall of the Exactech Optetrak Patella implant device.
The voluntary Exactech recall concerns Patella lots that were packaged without the ethylene vinyl alcohol (EVOH) layer. Experts advised doctors not to implant these potentially harmful devices packaged in faulty packaging.
Medical professionals were advised to stop using the product immediately and set aside any that might be affected. They were instructed that healthcare professionals should report any adverse reactions or noticeable quality issues as patients come forward.
Our attorneys handle Exactech recall lawsuits nationwide. If you’ve experienced failure of an Exactech knee, hip, or ankle implant that was recalled, leading to revision surgery, we’re here to help.
Stay informed with the latest updates and insights on this page.
Don’t overlook your rights—contact our product liability lawyers at 800-967-8529 or online. You won’t incur any fees unless you receive financial compensation.
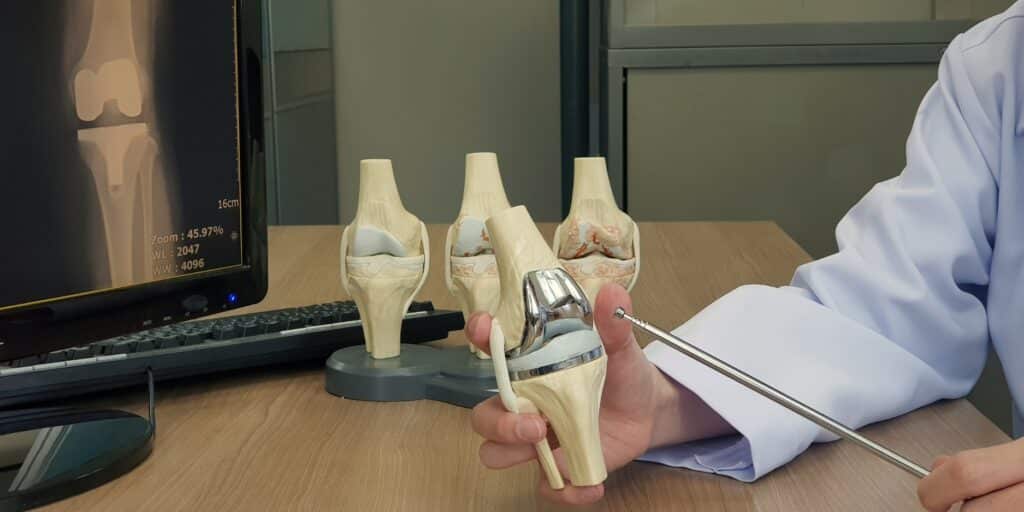
Exachtech Addresses Oxidation Risk with Packaging Recall
From 2004 to August 2021, Exachtech used two types of packaging materials in its process: (1) Low-Density Polyethylene (LDPE), Nylon, and EVOH, or (2) LDPE and Nylon without EVOH.
EVOH improves the prevention of oxygen permeation. Thousands of devices were packaged without using an EVOH oxygen barrier layer, which potentially causes unintended oxidation and early degradation of the affected device.
Given the potential for oxidation-related issues, Exachtech voluntarily recalled these lots as a precautionary measure.
The recall emphasizes the importance of surgeons regularly monitoring patients with affected devices for signs of wear and failure. The crucial issue is determining how many thousands of patients might suffer from device-related pain or failure, potentially needing revision surgery.
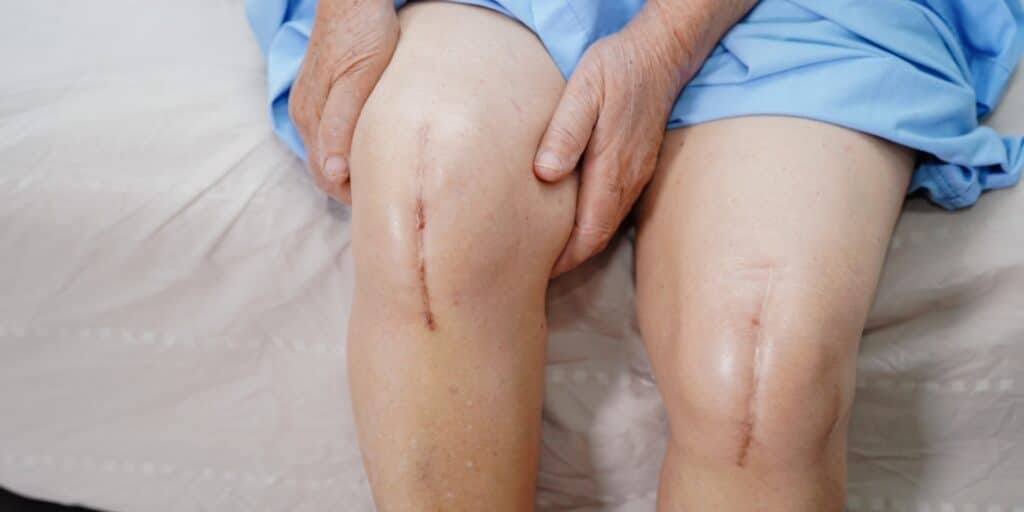
Stay Informed on Patella Device Recall and Risks
The lot-specific voluntary recall relates to Patella devices manufactured from 2004 through August 2021. These devices were marketed as Optetrak and cleared through 510(k): K932690, K933610, and K160484.
All healthcare professionals were instructed to review the following information and take any necessary actions. Then, the information advised diagnostic considerations, such as performing X-rays to evaluate the patient further if device failure is suspected.
Potential issues due to oxidation include:
– Accelerated device wear or failure
– Component cracking or fracture
– New or worsening pain
– Bone loss
– Swelling in the affected area
Key Information on Optetrak Device Failures
The recall names all of these knee replacement devices, which could necessitate revision surgery for patients.
Part Number Device Description Device Identifier
200-02-26 THREE PEG PATELLA 26MM 10885862039576
200-02-29 THREE PEG PATELLA 29MM 10885862039583
200-02-32 THREE PEG PATELLA 32MM 10885862039590
200-02-35 THREE PEG PATELLA 35MM 10885862039606
200-02-38 THREE PEG PATELLA 38MM 10885862039613
200-02-41 THREE PEG PATELLA 41MM 10885862039620
200-03-26 ONE PEG PATELLA 26MM 10885862039637
200-03-29 ONE PEG PATELLA 29MM 10885862039644
200-03-32 ONE PEG PATELLA 32MM 10885862039651
200-03-35 ONE PEG PATELLA 35MM 10885862039668
200-03-38 ONE PEG PATELLA 38MM 10885862039675
200-03-41 ONE PEG PATELLA 41MM 10885862039682
200-05-23 INSET PATELLA 23MM 10885862039835
200-05-26 INSET PATELLA 26MM 10885862039842
200-05-29 INSET PATELLA 29MM 10885862039859
200-07-26 ADVANCED PATELLA 26MM 3 PEG IMPLANT 10885862314260
200-07-29 ADVANCED PATELLA 29M 3 PEG IMPLANT 10885862314277
200-07-32 ADVANCED PATELLA 32MM 3 PEG IMPLANT 10885862314284
200-07-35 ADVANCED PATELLA 35MM 3 PEG IMPLANT 10885862314291
200-07-38 ADVANCED PATELLA 38MM 3 PEG IMPLANT 10885862314307
The Hidden Dangers of 510(k) Fast-Track FDA Approvals
Between 1994 and 2017, Exactech secured multiple 501(k) clearances from the FDA for its Optetrak, Optetrak Logic, and Truliant total knee replacement implant systems and components.
The FDA’s 510(k) clearance, known as “fast-track” approval, does not require manufacturers to prove a product’s safety and effectiveness.
This process, known as “premarket notification” or 510(k) clearance, only requires the manufacturer to demonstrate that the device is substantially equivalent to a pre-MDA predicate device.
Consequentially, the FDA can “clear” a new device for sale in the USA. Under the 510(k) process, the component parts used in the Exactech Optetrak Devices were either approved by the FDA or marketed without receiving either 510(k) clearance or full approval by the FDA.
Simply put, not enough product development and testing was done to ensure product safety. This can lead to patient harm and potential recalls, which is precisely what has happened.
Protect Your Rights Against Defective Medical Products
At Yost Legal Group, our priority is ensuring patient safety and achieving effective outcomes for patients with life-altering defective medical products. Our knee replacement lawsuit attorneys will help you file a lawsuit to seek compensation.
Collaborative efforts are essential for the success of actions against medical device manufacturers—especially those who continuously market harmful products.
Our law firm is dedicated to defending the rights of individuals against powerful corporations. Accordingly, when huge medical device manufacturers like Exactech continue to put profits before people, we step in to level the playing field.
Learn about your legal rights. We can file a product liability case on your behalf if you have a defective medical device. The Yost Legal Group is filing the current personal injury lawsuits:
- Knee replacement recall
- Shoulder replacement recall
- Hip replacement recall
- Exactech lawsuits
Empowering Patients, Challenging Corporations
If you or a loved one had knee replacement surgery in the past ten years and are experiencing pain, a clicking noise, swelling, or any other medical concerns, contact your doctor immediately.
Contact The Yost Legal Group for a free and confidential consultation at 1-800-967-8529. Our personal injury law firm is committed to promptly and transparently addressing potential concerns. If you had knee replacement surgery and are affected by the Exactech knee recall, please contact us.
Hold Exactech accountable for the harm they’ve caused. Our legal team is committed to seeking justice and ensuring they change their ways. A product defect lawyer will file an Exacteck lawsuit on your behalf.
For more information or to discuss your case, call us at 1-800-967-8529 or click this link to request an appointment.
By pursuing an Exactech knee replacement lawsuit, you will help send a powerful message for change. You may qualify for a significant financial recovery.
At The Yost Legal Group, you never have to pay anything upfront. We only get paid after we settle your case. If there is no recovery, no legal fees or expenses are due. Our personal injury law attorneys will provide legal representation with a free case evaluation.
_____________________________________________________________
Consumers are encouraged to report any adverse events or quality concerns related to this recall to the FDA through the MedWatch reporting system. You can go online at the FDA Medwatch Website or call 1-800-FDA-0178.